
M. Canevelli1, G. Bruno1, M. Valletta1, M. Cesari2,3
1. Department of Human Neuroscience, Sapienza University, Rome, Italy; 2. Geriatric Unit, IRCCS Istituti Clinici Scientifici Maugeri, Milan, Italy; 3. Department of Clinical Sciences and Community Health, University of Milan, Milan, Italy
Corresponding Author: Marco Canevelli, MD, PhD, Department of Human Neuroscience, Sapienza University of Rome, Viale dell’Università 30, 00185 Rome, Italy, Tel/fax +39 (0)6 49914604; Email: marco.canevelli@gmail.com
J Frailty Aging 2022;11(3)248-249
Published online May 31, 2022, http://dx.doi.org/10.14283/jfa.2022.43
The literature has frequently reported and discussed a relevant mismatch between the neuropathological modifications and the clinical manifestations of Alzheimer’s disease (AD) (1, 2). The dissociation between lesions and phenotypic expression has triggered a growing body of research, primarily because the discrepancy might pave the way for developing novel therapeutic opportunities. In particular, understanding the so-called “resilience to AD neuropathological changes” (3) (i.e., why a subset of individuals with AD brain lesions remain clinically unaffected) could provide insights into the critical mechanisms of brain damage in AD.
To date, research efforts in the field have tentatively explored the putative neurobiological underpinnings of the resilient response to AD, mainly focusing on beta-amyloid, tau, and neuroinflammation (3). However, other aging-related pathophysiological processes may likely influence the reserves against plaques and tangles beyond the brain responses and adaptations. In this regard, recent studies have shown that frailty, a condition of compromised homeostasis and increased vulnerability to stressors (4, 5), may contribute to the individual’s ability to cope with AD lesions, besides representing an independent risk factor for cognitive decline and dementia (6–8). In secondary analyses from the Rush Memory and Aging Project (MAP, a cohort study of older adults without dementia at baseline), 19% of participants exhibited an AD neuropathological burden (i.e., neurofibrillary tangles and amyloid plaques) that did not correspond with the expected cognitive impairment (9). Such dissociation between biological and clinical aspects seemed strongly influenced by frailty. In particular:
• Among participants expressing a high burden of AD pathology, only 17.1% of patients with frailty were dementia-free at the time of the death, in contrast with 42.8% of those with a low degree of frailty;
• Among those with a low neuropathological burden, dementia was more prevalent in those with frailty than robust ones (69% vs. 5%).
The reported findings were confirmed in subsequent analyses that considered a greater variety of neuropathological modifications, including hippocampal sclerosis, Lewy’s body pathology, TDP-43 pathology, arteriolosclerosis, and amyloid angiopathy (10). A similar picture emerges when the AD pathological changes are documented in vivo using neuroimaging and CSF biomarkers (11). In a cross-sectional analysis from the Alzheimer’s Disease Neuroimaging Initiative (ADNI), we recently showed that dementia was more prevalent in the frail (42.9%) than non-frail (18.7%) participants with abnormal biomarkers values (i.e., high florbetapir uptake at the amyloid PET scan). Altogether, these studies suggest that the degree of frailty influences the ability to tolerate the accrual of neuropathological lesions, potentially affecting their phenotypic expression (Figure 1). Robust individuals who have accumulated fewer age-related health deficits may present higher reserves in the face of neurodegeneration. On the contrary, even a minimal burden of brain lesions may result in overt cognitive impairment when frailty is present.
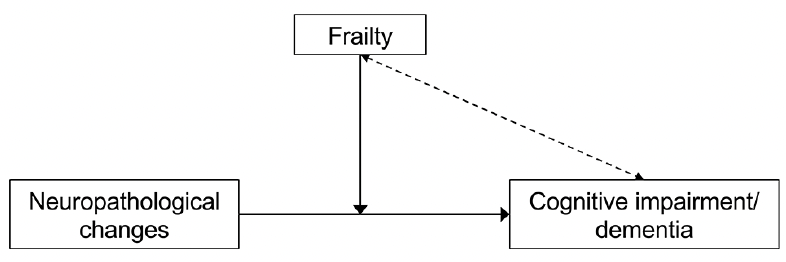
Figure 1. The role of frailty in the relationship between neuropathological changes and clinical manifestations of Alzheimer’s disease
Frailty moderates the association between Alzheimer’s disease neuropathological modifications and clinical manifestations (solid line), besides constituting an independent risk factor for cognitive impairment and dementia (dotted line).
These observations may contribute to a gradual shift in the notion of sporadic AD and dementia, which are increasingly conceived as multidimensional age-related conditions rather than discrete brain diseases with specific pathological hallmarks (9, 10). Accordingly, the study of resilience (or excess of vulnerability) to AD might be improved by exploring the multisystemic and interacting mechanisms that shape the homeostatic reserves in the context of the aging process. Under this scenario, the investigation of frailty may promote essential advancements in the field. More research is needed to understand the potential role of frailty as a latent factor in the relationship between neuropathology and phenotypic manifestations in AD. Beyond speculative purposes, this may have important clinical implications. Indeed, measuring frailty might provide prognostic information on the likelihood of developing dementia in the growing population of subjects that are tested with candidate AD biomarkers. At the same time, reducing frailty (i.e., maintaining or improving general health) might theoretically mitigate the risk of dementia in at-risk individuals.
Competing interests: Authors have no competing interest to disclose for the present study. Matteo Cesari has received honoraria for presentations at scientific meetings and served as a member of Scientific Advisory Boards for Nestlé Health Science.
References
1. Boyle PA, Wilson RS, Yu L, Barr AM, Honer WG, Schneider JA, et al. Much of late life cognitive decline is not due to common neurodegenerative pathologies. Ann Neurol. 2013 Sep;74(3):478–89.
2. Espay AJ, Vizcarra JA, Marsili L, Lang AE, Simon DK, Merola A, et al. Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer diseases. Neurology. 2019 12;92(7):329–37.
3. Gómez-Isla T, Frosch MP. Lesions without symptoms: understanding resilience to Alzheimer disease neuropathological changes. Nat Rev Neurol. 2022 Mar 24;
4. Howlett SE, Rutenberg AD, Rockwood K. The degree of frailty as a translational measure of health in aging. Nat Aging. 2021 Aug;1(8):651–65.
5. Rockwood K, Mitnitski A. How Might Deficit Accumulation Give Rise to Frailty? J Frailty Aging. 2012;1(1):8–12.
6. Ward DD, Ranson JM, Wallace LMK, Llewellyn DJ, Rockwood K. Frailty, lifestyle, genetics and dementia risk. J Neurol Neurosurg Psychiatry. 2022 Apr;93(4):343–50.
7. Ward DD, Wallace LMK, Rockwood K. Cumulative health deficits, APOE genotype, and risk for later-life mild cognitive impairment and dementia. J Neurol Neurosurg Psychiatry. 2021 Feb;92(2):136–42.
8. Houles M, Canevelli M, van Kan GA, Ousset PJ, Cesari M, Vellas B. Frailty and Cognition. J Frailty Aging. 2012;1(2):56–63.
9. Wallace LMK, Theou O, Godin J, Andrew MK, Bennett DA, Rockwood K. Investigation of frailty as a moderator of the relationship between neuropathology and dementia in Alzheimer’s disease: a cross-sectional analysis of data from the Rush Memory and Aging Project. Lancet Neurol. 2019;18(2):177–84.
10. Wallace LMK, Theou O, Darvesh S, Bennett DA, Buchman AS, Andrew MK, et al. Neuropathologic burden and the degree of frailty in relation to global cognition and dementia. Neurology. 2020 Dec 15;95(24):e3269–79.
11. Canevelli M, Arisi I, Bacigalupo I, Arighi A, Galimberti D, Vanacore N, et al. Biomarkers and phenotypic expression in Alzheimer’s disease: exploring the contribution of frailty in the Alzheimer’s Disease Neuroimaging Initiative. Geroscience. 2021 Apr;43(2):1039–51.